DÜNNDARMZOTTEN MIT BAKTERIEN
Grundlagen des Mikrobioms
Grundlagen des Mikrobioms

Bestimmung des Mikrobioms
In der Vergangenheit wurden Mikroorganismen mittels kulturbasierter Methoden bestimmt. Diese Methoden waren mit erheblichen Einschränkungen verbunden, wie z. B. der Tatsache, dass einige Organismen des Mikrobioms nicht kultiviert werden können.1-3
Nicht alle Mikroorganismen sind unter den In-vitro-Bedingungen in kulturbasierten Methoden lebensfähig.1
Bei den molekularen Methoden der nächsten Generation werden bakterielle DNA-Sequenzen anstelle von Organismen verwendet, um die Taxonomie, die relative Häufigkeit und die Funktion zu bestimmen.1,4-6 Im Folgenden werden kurz die am häufigsten verwendeten Methoden beschrieben.
Überblick über gängige Methoden zur Bestimmung des Mikrobioms
Amplicon-basierte Sequenzierung von Markergenen
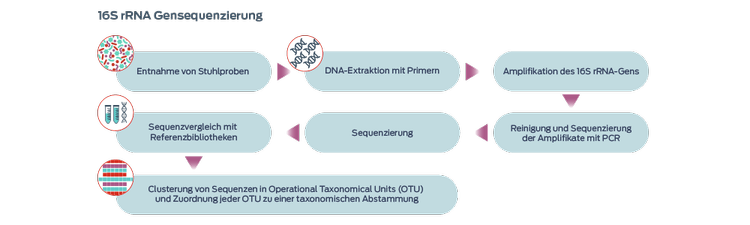
Der am häufigsten verwendete Ansatz ist die Sequenzierung des 16S-ribosomalen RNA-Gens (rRNA), das in allen Bakterien und Archaeen, jedoch nicht in Pilzen oder im Tierreich vorkommt. Ziel dieses Ansatzes ist es, eine Zählung der Mikroben in der Probe durchzuführen.1,4,7 Die Identifizierung des Mykobioms ersetzt die Sequenzierung der 18S-rRNA-Gene.7
Das 16S-rRNA-Gen ist bei allen Bakterienarten ein hoch konserviertes Gen und enthält hypervariable Regionen, die eine Differenzierung ermöglichen.1 Dieser Ansatz erlaubt die Identifizierung vieler Mikroorganismen aus einer Probe, einschließlich Mikroorganismen, die nicht erfolgreich kultiviert werden können.1 Diese Technik unterscheidet jedoch nicht zwischen lebenden oder toten, transienten oder residenten, kommensalen oder pathogenen bzw. antibiotikaresistenten oder -empfindlichen Mikroorganismen.1
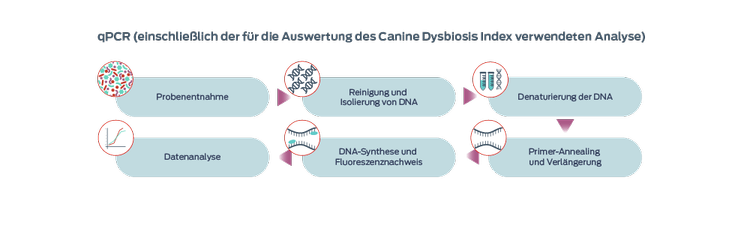
Quantitative Polymerase-Kettenreaktion (qPCR)
Die quantitative PCR ist eine schnelle und kostengünstige Methode zur Quantifizierung der interessierenden spezifischen Bakterientaxa und ermöglicht die Festlegung von Referenzintervallen. Auf Grundlage der qPCR an sieben Bakterienarten wurde der Canine Microbiota Dysbiosis Index entwickelt, mit dem Veränderungen im Mikrobiom als Reaktion auf eine Therapie nachverfolgt werden können.3
Dies ist ein Link zu einem Artikel auf Englisch, der nicht auf Deutsch verfügbar ist.
Beschreibung der „Mikrobenzählung“
Operational Taxonomic Units (OTUs) sind Cluster von DNA-Sequenzen, die auf Ähnlichkeit basieren. Zur Identifizierung der Mikroben eines Clusters werden repräsentative Sequenzen aus jeder OTU mit Referenzdatenbanken verglichen.
Die Alpha-Diversität und die Beta-Diversität geben Auskunft über die Arten-Bandbreite im Mikrobiom. Die Alpha-Diversität ist ein Maß für die Artenvielfalt in einer Probe.1 Die Alpha-Diversität drückt die Häufigkeit (die Anzahl der Arten/OTUs und deren Verteilung) und die Gleichmäßigkeit (die relative Häufigkeit der verschiedenen OTUs/Arten und deren Verteilung) aus. Sie basiert auf Algorithmen und wird üblicherweise als Index wie beispielsweise Shannon-Index oder Simpson-Index ausgedrückt.1 Die Beta-Diversität ist ein Maß für die Verschiedenheit zwischen den Proben, die häufig als Clustermuster dargestellt wird. Zu den Systemen, die zur Berechnung der Beta-Diversität verwendet werden, gehören der Bray-Curtis-Index und der ungewichtete oder gewichtete UniFrac-Abstand.1
Von der Zählung zur Funktion
Mit zunehmender Erforschung des Mikrobioms kristallisieren sich zwei zentrale Fragen heraus: Welche Mikroben liegen vor und welche Funktion üben sie aus? Die Identifizierung der im Mikrobiom vorhandenen Mikroorganismen gibt keinen Aufschluss über ihre Funktion.3,5 Es ist wahrscheinlich, dass bestimmte metabolische und molekulare Funktionen von mehr als einer Mikrobe ausgeführt werden, was zu einem redundanten Ökosystem führt, das sich durch Flexibilität und Resilienz auszeichnet.3,8
Aufgrund dieser Redundanz reicht eine Analyse des Mikrobioms basierend auf den in der Population vorhandenen Mikrobenarten nicht aus, um funktionelle Veränderungen im Mikrobiom zu erkennen. Durch eine Intervention oder eine bestimmte Bedingung kann die relative Häufigkeit der Mikroben potenziell verändert werden, doch das betreffende Mikrobiom kann die mikrobiellen Aktivitäten und den Stoffwechsel so verschieben, dass diese Veränderung kompensiert wird. Zur Erkennung dieser Verschiebungen bedarf es verschiedener Analyseverfahren.
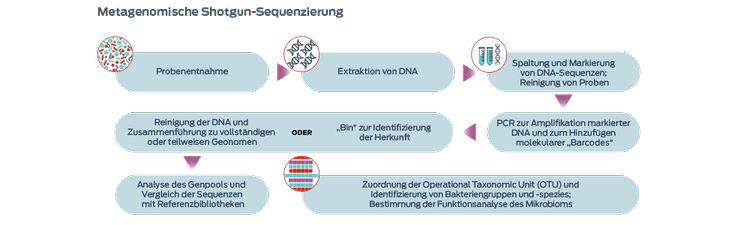
Metagenomische Schrotschuss-Sequenzierung
Die sogenannten Schrotschuss (Shotgun)-Methoden tragen diesen Namen, weil sie nicht zielgerichtet sind (d. h., sie zielen nicht auf den Nachweis einer bestimmten Gruppe von Mikroben ab). Sie gewinnen im Rahmen der Analyse des Mikrobioms von Haustieren zunehmend an Bedeutung, da sie den Vorteil der Sequenzierung funktioneller Gene anstelle der bloßen Identifizierung von Bakterien bieten.3-5 Für diese Verfahren sind jedoch größere Mengen an DNA aus den Proben erforderlich, und sie sind außerdem kostenintensiver.3
Die Long-Read-Sequenzierung ermöglicht die Zusammensetzung vollständiger Genome, einschließlich der Gene, die bei der Short-Read-Metagenomik normalerweise übersehen werden und zusätzliche biologische Erkenntnisse liefern.6
Metabolomik, Proteomik und Transkriptomik
Bei diesen Verfahren wird die funktionelle Aktivität eines Mikrobioms gemessen.4 Metabolomische Ansätze kommen bei der Bestimmung des Metabolitenprofils zum Einsatz und werden in der Regel mit Verfahren wie Kernspinresonanz, Spektroskopie, Massenspektroskopie und Flüssigkeitschromatographie durchgeführt.7 Mit diesen Ansätzen werden die von Mikroben regulierten Signalwege untersucht, wie die Herstellung kurzkettiger Fettsäuren, der Gallensäurestoffwechsel, die Produktion von Neutrotransmittern und die Indolproduktion.3

Weitere Bereiche des Microbiome Forum durchsuchen



Weitere Informationen
- Belas, A., Marques, C., & Pomba, C. (2020). The gut microbiome and antimicrobial resistance in companion animals. In Duarte, A. & Lopes da Costa, L. (Eds.), Advances in Animal Health, Medicine and Production (1st ed.), pp. 233—245. Springer International Publishing
- Cunningham, M., Azcarate-Peril, M. A., Barnard, A., Benoit, V., Grimaldi, R., Guyonnet, D.,…Gibson, G. R. (2021). Shaping the future of probiotics and prebiotics. Trends in Microbiology, 29(8), 667—685. doi:10.1016/j.tim.2021.01.003
- Pilla, R., & Suchodolski, J. S. (2021). The gut microbiome of dogs and cats, and the influence of diet. Veterinary Clinics of North America Small Animal Practice, 51(3), 605–621. doi:10.1016/j.cvsm.2021.01.002
- Bokulich, N. A., Ziemski, M., Robeson, M. S., & Kaehler, B. D. (2020). Measuring the microbiome: Best practices for developing and benchmarking microbiomics methods. Computational and Structural Biotechnology Journal, 18, 4048–4062. doi:10.1016/j.csbj.2020.11.049
- Radjabzadeh, D., Uitterlinden, A. G., & Kraaij, R. (2017). Microbiome measurement: Possibilities and pitfalls. Best Practice & Research Clinical Gastroenterology, 31, 619–623. doi:10.1016/j.bpg.2017.10.008
- Cusco, A., Pérez, D., Viñes, J., Fàbregas, N., & Francino, O. (2020). Long-read metagenomics retrieve complete single-contig bacterial genomes from canine feces. In review, BMC Genomics. doi:10.21203/rs.3.rs-135952/v1
- Marchesi, J. R. & Ravel, J. (2015). The vocabulary of microbiome research: a proposal. Microbiome, 3, 31. doi:10.1186/s40168-015-0095-5
- Koidl, L., & Untersmayr, E. (2021). The clinical implications of the microbiome in the development of allergy diseases. Expert Review of Clinical Immunology, 17, 115—126. doi:10.1080/1744666X.2021.1874353